La encefalomiopatía es un trastorno genético neurometabólico poco frecuente, multisistémico y progresivo. Se caracteriza por una disfunción mitocondrial, que son los orgánulos celulares encargados de producir energía. Esta disfunción provoca una serie de síntomas que afectan a diversos órganos y sistemas, siendo la encefalomiopatía (afectación del cerebro y los músculos), la acidosis láctica y los episodios que simulan un ictus (stroke-like) las manifestaciones más comunes.
Epidemiología
La prevalencia e incidencia exactas de la encefalomiopatía son desconocidas. Se estima que afecta a 1 de cada 000 personas, siendo uno de los trastornos mitocondriales más frecuentes. No hay diferencias significativas en la prevalencia entre sexos.
Descripción Clínica
Las encefalomiopatías son enfermedades multiorgánicas con manifestaciones clínicas variables. Los síntomas suelen aparecer antes de los 20 años de edad, aunque pueden desarrollarse a cualquier edad. La característica distintiva son los episodios parecidos a un ictus, que causan vómitos, cefalea o epilepsia, seguidos de pérdida de conciencia y, a menudo, hemiparesia (debilidad en un lado del cuerpo), hemianopsia (pérdida de la visión en la mitad del campo visual) y ceguera cortical. Los pacientes pueden presentar antecedentes de retraso del desarrollo, talla baja o problemas de aprendizaje previos a estos episodios. Otras manifestaciones comunes incluyen:
- Acidosis láctica: Un exceso de ácido láctico en la sangre, lo que indica una disfunción mitocondrial.
- Miopatía: Debilidad muscular, hipotonía (tono muscular bajo) e intolerancia al ejercicio.
- Endocrinopatía: Afectación de las glándulas endocrinas, como la diabetes (principalmente tipo 2, y ocasionalmente tipo 1) o problemas de la tiroides.
- Polineuropatía: Afectación de los nervios periféricos, causando debilidad, entumecimiento y hormigueo en las extremidades.
- Miocardiopatía: Afectación del músculo cardíaco, que puede llevar a insuficiencia cardíaca.
- Hipoacusia neurosensorial: Pérdida de la audición debido a problemas en el oído interno.
- Pseudoobstrucción intestinal: Dificultad para el paso de los alimentos por el intestino.
- Manifestaciones psiquiátricas: Pérdida de apetito, depresión, ansiedad, psicosis, trastorno bipolar, demencia, trastornos del espectro autista y, en raras ocasiones, trastornos de la conducta.
- Síndrome nefrótico: Afectación de los riñones, causando proteinuria (proteína en la orina).
- Afectación dermatológica: Púrpura (manchas moradas en la piel), hirsutismo (exceso de vello) y eritema (enrojecimiento de la piel).
El curso de la encefalomiopatía suele ser progresivo, con deterioro cognitivo gradual, discapacidad y fallecimiento precoz.
Etiología
La patogenia exacta de la encefalomiopatía no se conoce completamente. Se sabe que el agotamiento de la energía debido a la disfunción mitocondrial subyace a las manifestaciones clínicas. Se han identificado mutaciones en 19 genes mitocondriales, siendo la más frecuente la mutación 3243A>G en el gen mitocondrial del tRNA Leu (UUR) (MT-TL1), presente en aproximadamente el 80% de los casos. Estas mutaciones conducen a una disfunción del ARNt mitocondrial, provocando alteraciones en la síntesis proteica mitocondrial.
Métodos Diagnósticos
El diagnóstico de la encefalomiopatía se basa en una combinación de los rasgos clínicos característicos, hallazgos de laboratorio indicativos de disfunción mitocondrial y pruebas genéticas. Los hallazgos de pruebas diagnósticas incluyen:
- Acidosis láctica: Confirma la disfunción mitocondrial.
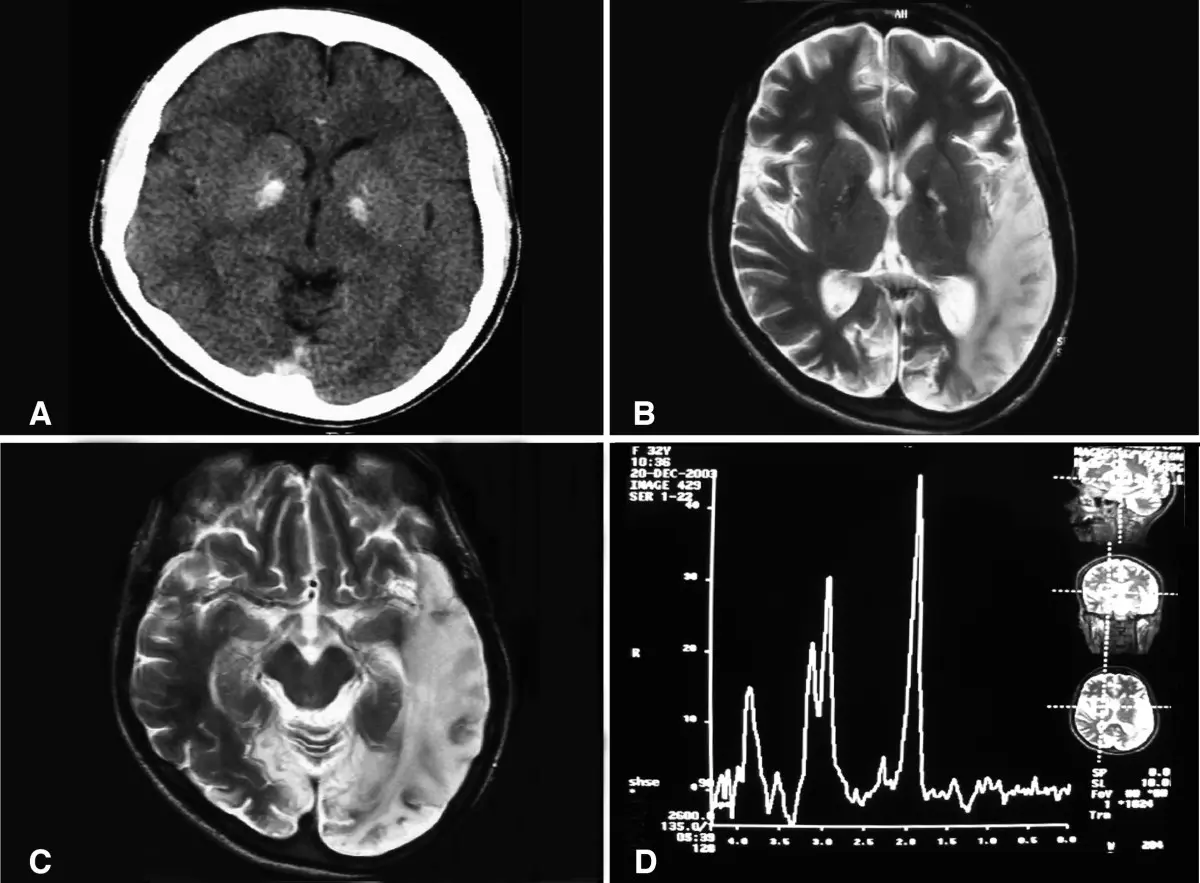
- Imágenes cerebrales: Durante el accidente cerebrovascular muestran alteraciones que no se ajustan al territorio vascular.
- Biopsia muscular: Muestra fibras rojas rasgadas, típicas de la encefalomiopatía .
- Análisis de la cadena respiratoria: Muestra múltiples defectos parciales en la cadena respiratoria mitocondrial.
- Pruebas genéticas: Confirman la mutación en el ADN mitocondrial.
Es importante destacar que los resultados de las pruebas genéticas por sí solos no pueden predecir el diagnóstico, debido a la variabilidad de los fenotipos causados por la mutación (m.3243A-a-G), que van desde la encefalomiopatía grave hasta diabetes, sordera o incluso pacientes asintomáticos.
Diagnóstico Diferencial
Es necesario distinguir la encefalomiopatía de otras enfermedades que causan un accidente cerebrovascular isquémico agudo de aparición juvenil. En pacientes con migraña, epilepsia o encefalitis, se debe descartar la encefalomiopatía. Otros diagnósticos diferenciales incluyen otros trastornos mitocondriales que comparten manifestaciones clínicas con la encefalomiopatía.
Diagnóstico Prenatal
El diagnóstico prenatal, aunque posible, es difícil debido a la heterogeneidad en la proporción de mutaciones en los distintos tejidos.
Consejo Genético
La encefalomiopatía es un trastorno de herencia mitocondrial por vía materna y rara vez por una mutación de novo. El asesoramiento genético es complejo debido a la heteroplasmia, la presencia de diferentes proporciones de ADN mitocondrial mutado y normal en diferentes tejidos.
Manejo y Tratamiento
Actualmente no existe un tratamiento específico para la encefalomiopatía. Los agentes que se han utilizado, aunque con evidencia insuficiente, incluyen:
- Arginina
- Taurina
- Citrulina
- Carnitina
- Creatina monohidrato
- Idebenona
- Dicloroacetato
- Coenzima Q10
- Menadiona
- Filoquinona
- Ácido ascórbico
- Riboflavina
- Nicotinamida
Las manifestaciones endocrinas, cardíacas, gastrointestinales, psiquiátricas, renales y dermatológicas se tratan con medidas sintomáticas estándar. La encefalomiopatía progresiva debe monitorizarse estrechamente.

Pronóstico
La encefalomiopatía progresa a lo largo de los años con un acúmulo de déficits neurológicos y presenta una elevada morbimortalidad.
Si quieres conocer otros artículos parecidos a Encefalomiopatía: un trastorno multisistémico progresivo puedes visitar la categoría Salud.